주문제작 서비스 문의
찾으시는 제품이 저희 사이트에 없으신가요? 단백질 주문 제작 서비스를 이용해보세요!
주문제작 서비스 문의 >>





















 Limited Edition Golden Llama is here! Check out how you can get one.
Limited Edition Golden Llama is here! Check out how you can get one.  Limited Edition Golden Llama is here! Check out how you can get one.
Limited Edition Golden Llama is here! Check out how you can get one.
Offering SPR-BLI Services - Proteins provided for free! Offering SPR-BLI Services - Proteins provided for free!
Here come GMP Grade Cytokines!Free Sample is available! Here come GMP Grade Cytokines!Free Sample is available!

최근 몇 년간 바이오 제약 산업은 지속적인 기술 발전을 거듭했으며, 여러 분야 중 세포 유전자 치료제가 괄목할 만한 성장을 거두었습니다. 차세대 의약품에서 생물학적 제제가 차지하는 비중이 커짐에 따라 이러한 생물학적 의약품의 안전성을 보장하는 것이 최우선 과제가 되었습니다. 따라서 세포 유전자 완제뿐만 아니라 제조 및 생산 과정에서 사용되는 원료 물질에 대해서도 더욱 면밀한 조사가 이루어지고 있습니다. 더욱이, FDA가 '최고 품질'의 적합한 원료를 사용할 것을 강조하면서 이에 대한 중요성은 더욱 부각되고 있습니다. 마찬가지로, International Pharmaceutical Regulatory Plan Cell Therapy Working Group은 사람에게 투여될 CGT 제품의 제조 및 허가 시 고품질 원료 물질을 사용하는 것에 대한 심의 문서 초안을 작성하며 중요성을 강조했습니다.
규제 기관이 '고품질' 원료 물질 사용을 강조하는 상황에서 세포 치료제 제조업체에게 이는 어떤 의미일까요?
간단히 말해, 세포 치료제 제조업체는 공급업체에서 공급받거나 자체 개발한 원료 물질의 품질을 보장하기 위해 반드시 실사를 수행해야 합니다.
사이토카인 및 기타 성장 인자를 포함한 관련 원료 물질의 품질 관리 방법 및 표준에 대한 평가는 임상시험을 진행하기 전에 임상시험용 신약(investigational new drug, IND) 허가 신청 시 필수적으로 거쳐야 하는 단계입니다. 약전에 명시된 방법은 USP 또는 유럽 의약품 및 의료 서비스 품질 위원회(European Directorate for the Quality of Medicines & Healthcare, EDQM)와 같은 기관에서 명시한 바를 기반으로 하며, 제조 및 출하에 대한 기본 요건이 설정되어 있는 표준화된 접근 방식입니다. International Conference on Harmonization (ICH) 문서와 같이 약전에 명시되어 있지 않은 방법은 제품 및 제조 공정의 특정 특성을 평가하기 위해 맞춤화된 보조적인 분석 접근법입니다.
현재 많은 상용 제품이 이러한 표준을 준수하여 시판되고 있으며, 이러한 제품은 일반적으로 'GMP 등급' 또는 'cGMP 등급'이라고 라벨에 기재되어 있습니다. 그러나 명시된 표준을 준수하는 제품은 자체 규제 대상인 경우가 많습니다. 따라서 세포 치료제 제조업체는 여전히 실사를 수행해야 할 책임이 있으며, 이로 인해 신뢰할 수 있는 공급업체를 찾기가 어려울 수 있습니다.
본 인사이트에서는 IND 요건을 충족하고 임상 단계로 성공적으로 진입하는 데 중요한 GMP 등급 원료 물질 품질 관리 시스템의 세 가지 주요 측면을 강조하고자 합니다.
원료 물질 안전성 평가는 다양한 오염물질 관리 및 검출 전략을 통해 최종 치료제에 해를 끼치거나 영향을 미칠 수 있는 잔류물을 제한하는 것으로 정의됩니다. 여기에는 멸균 시험, 내독소, 마이코플라즈마, 잔류 숙주 세포 DNA, 잔류 숙주 세포 단백질과 같은 오염물질 평가가 포함됩니다.
• USP<71>멸균 시험(Sterility Tests)
최종 세포 및 유전자 치료제의 멸균 상태를 보장하는 것은 까다로운 작업입니다. 최종 치료제는 세포이기 때문에 멸균 여과, 고압 멸균 등 쉽게 적용할 수 있는 방법이 없습니다. 따라서 제조업체는 최종 제품의 무균성을 보장하기 위해 제조 공정 전반에 걸쳐 매우 주의를 기울여야 합니다. 이는 사용되는 원료 물질에도 적용되므로 제조업체는 공급업체를 엄격하게 평가해야 합니다. 그러므로 당사를 포함한 GMP 등급 제조업체의 원료 물질 공급업체는 B+A 청절실 내 자동 충전-마감, 온라인 환경 모니터, USP 준수 멸균 시험을 도입하여 사이토카인 및 기타 성장 인자를 엄격하게 관리해야 합니다. 따라서 원료의 멸균성을 평가하는 것은 최종 치료제의 멸균 상태를 보장하는 주요 방법입니다. 자세한 내용은GMP 제품 품질에 대한 심층 해석에 관한 특별 주제 - 주제 2를 참조하시기 바랍니다.
• USP<85>내독소 시험(Endotoxin Tests)
내독소는 박테리아 세포 내에 존재하는 독소로, 세포가 분해된 후 방출되어 보툴리누스 중독과 같은 질병을 유발할 수 있습니다. 생물학적 제제의 내독소 수치를 낮게 유지하는 것은 환자의 안전을 보장하고 내독소 관련 질병을 예방하는 데 필수적입니다. 이를 위해 여러 가지 방법을 사용할 수 있으며, 정성적 겔화 및 발색 LAL 기반 방법이 이에 해당합니다. 그러나 일관되지 않거나 상충되는 결과가 있는 경우 겔화법의 결과가 신뢰할 수 있는 것으로 간주됩니다. 내부적으로는 표 1에 표시된 시료 결과의 정확성을 보장하기 위해 USP 참조 표준 내독소 보정 물질과 비교하여 보정한 LAL 방법을 활용합니다. 그 후, 내독소 시험 방법이 유효한지 확인하기 위해 USP<85>에 명시된 대로 예비 시험을 수행했으며, 허용 가능한 내독소 회수율은 50~200% 범위 내로 정의했습니다.
표 1. 내독소 시료 곡선 평가
| Theoretical value | Detected value | Recovery rate |
|---|---|---|
| 50 EU/ml | 55.04 EU/ml | 113.0% |
| 5 EU/ml | 5.96 EU/ml | 104.7% |
| 0.5 EU/ml | 0.6 EU/ml | 108.5% |
| 0.05 EU/ml | 0.06 EU/ml | 108.0% |
• USP<63>마이코플라즈마 시험(Mycoplasma Tests)
마이코플라즈마는 세포 및 조직 배양에서 흔히 발생하는 오염 물질로, 세포 성장과 표현형에 변화를 일으킵니다. 마이코플라즈마 시험은 생물학적 제제 및 그 원료 물질의 신뢰성을 보장하는 데 매우 중요합니다. 마이코플라즈마를 평가할 때는 고체 배지에서 정형성(typical) 마이코플라즈마 콜로니의 성장을 모니터링하는 배양 방법을 최적 기준으로 삼고 있습니다. 유효성이 확인된 다른 방법으로는 형광 염료를 사용하여 세포 표면의 특징적인 미립자 또는 필라멘트 패턴을 강조하는 방법이 있습니다. 핵산 증폭 기술이나 효소 활성 기반 방법도 사용할 수 있지만, 세포 배양에 대한 적절한 밸리데이션과 방법 간의 비교 또한 다루어야 합니다.
• USP<509, 1132="">잔류 숙주 세포 DNA 및 단백질(Residual Host Cell DNA and Proteins)
숙주 세포의 DNA와 단백질 또한 안전성 위험을 방지하기 위해 허용 가능한 수준으로 관리해야 하는 다른 인자에 해당합니다. 외부 DNA와 단백질은 면역원성 및 발암원성 반응을 일으켜 더 많은 해를 끼칠 수 있습니다. 모든 원료 물질 및 후속 제조 공정에서 이러한 불순물을 제거하는 것은 환자 안전에 매우 중요합니다.
물론, 모든 시험법 또는 분석법과 마찬가지로, 사용된 시험이 정확한지 확인하려면 밸리데이션이 필수적입니다. 위에 언급된 USP 문서는 생물학적 물질 전반에 걸쳐 표준화된 시험법을 평가하기 위한 분석 프로토콜과 명확한 기준을 제공합니다. 그러나 생체 활성과 같은 물질별 평가의 경우, 분석 방법에 대한 밸리데이션을 먼저 해야 하며, 이러한 밸리데이션을 수행할 때는 '분석 절차 텍스트 및 방법의 밸리데이션(Validation of Analytical Procedures Text and Methodology)'이라는 제목의 ICH Q2(개정 1)에 따르는 것이 가장 일반적입니다.
표 2. 고려해야 할 밸리데이션 특성
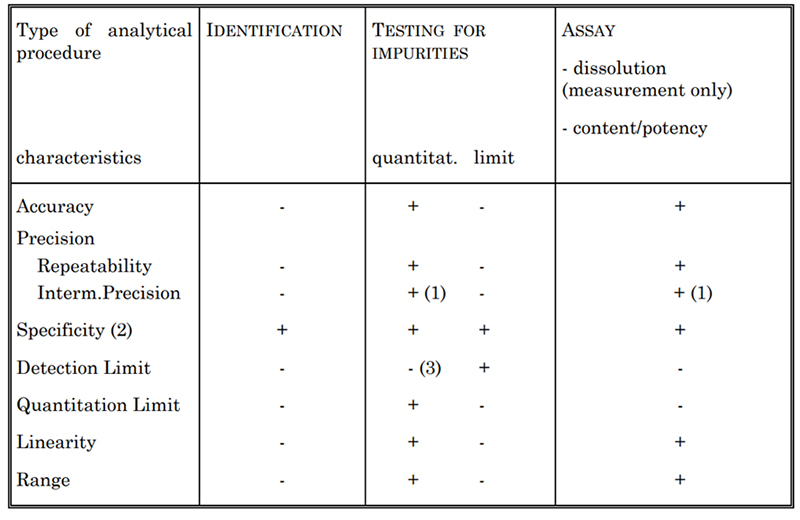
밸리데이션할 분석 유형에 따라서, 일반적으로 표 2에 나열된 대로 특정 특성을 평가합니다. 여기에는 특이도, 정확도, 정밀도, 검출 한계, 정량 한계, 선형성, 범위가 포함됩니다. 예를 들어, 당사는 사이토카인 농도를 측정하기 위해 자외선 분광 광도 분석법, Lowry 방법 등 다양한 분석법을 설정합니다. 이러한 각 방법은 USP 및 ICH Q2 가이드라인에 따라 완전히 밸리데이션 되었습니다.
표 3. 자외선 분광 광도 분석의 밸리데이션
| Method Validation Parameter | Result | Criteria | Conclusion |
|---|---|---|---|
| Accuracy | Recovery Rate: 96% | Recovery Rate: 90%-108% | Pass |
| Repeatability | RSD: 0.05% | RSD≤3% | Passtd> |
| Intermediate Precision | RSD: 0.48% | RSD≤3% | Pass |
| Robustness | RSD: 0.04% | RSD≤3% | Pass |
| Linearity | R2: 0.99925 | R2>0.999 | Pass |
| Range | 0.0452-0.452 mg/ml | 0.0452-0.452 mg/ml | Pass |
자외선 분광 광도 분석법을 추가로 밸리데이션하기 위해 2차 확인 방법인 Lowry 방법을 사용했습니다. 상당 기간에 걸쳐 확립되어 있고 잘 정립된 방법인 Lowry 방법은 비교를 위한 탄탄한 기반을 제공합니다. 따라서 서로 다른 스파이크 시료 6개의 정량적 결과를 직접 비교하면 표 4에 나타난 것처럼 UV 방법의 정확성과 신뢰성을 보장할 수 있습니다.
표 4. 2차 확인 분석 - Lowry 방법
| Method Validation | Lowry | UV Method | Conclusion |
|---|---|---|---|
| Quantitative Result | 461 ug/ml | 436 ug/ml | Pass |
| 476 ug/ml | 436 ug/ml | ||
| 465 ug/ml | 436 ug/ml | ||
| 472 ug/ml | 436 ug/ml | ||
| 468 ug/ml | 436 ug/ml | ||
| 461 ug/ml | 435 ug/ml |
여기에는 동일한 분석법 밸리데이션 프레임워크를 사용하며 세포 활성과 같은 다른 분석 또한 포함됩니다. 예를 들어, 당사의 TNF-α 분석에는 정확도, 중간 정밀도, 선형성, 범위가 포함됩니다. 그러나 분석 목적에 따라 기준이 다릅니다.
표 5 TNF-α 세포 활성 밸리데이션의 예시
| Method Validation Parameter | Result | Criteria | Conclusion |
|---|---|---|---|
| Relative Accuracy | Bias:1.1% Slope:1.01 | Bias within ±12% range & Slope of regression equation between 0.80 and 1.25 | Pass |
| Intermediate Precision | GCV*:11.2% | (GCV)* ≤ 20% | |
| Specificity | Difference % (buffer): 8.8% | Difference % (buffer) ≤ ±10% | |
| Linearity | Correlation Coefficient: 0.97 | Correlation Coefficient ≥ 0.95 | |
| Potency Range | Potency Range: 64% -156% | Range of product efficacy standards (64% -156%) |
*GCV는 표준 편차의 역로그 값으로 계산함. GCV = 역로그(SD)
안정성 시험은 제품의 유통기한을 평가하는 기초가 됩니다. 따라서 이 시험은 전체 약물 개발, 임상, 시장 출시 및 시판 후 모니터링 전반에 걸쳐 품질 연구 프로세스 목적으로 중요한 구성 요소입니다. 이는 제품의 고유한 품질과 특성을 기반으로 안정성 시험을 수행해야 함을 의미합니다. 또한 안정성 연구를 체계적으로 수행하여 로트 간 편차를 최소화하는 동시에 배송 시 보관 방법에 대한 명확한 지침을 제공합니다.
Arrhenius 식에 기반한 가속 안정성 시험 방법은 실행 가능한 시간 내에 안정성과 일관성을 평가하기 위해 사용하며 널리 알려진 전략으로, 최근 몇 년간 수많은 연구를 통해 그 적용 가능성과 정확성이 입증되었습니다. 유럽 표준화 위원회(European Committee for Standardization)에서 발행한 2002년 문서 'EN13640 생체 외 진단 의료 기기 안정성 시험(EN13640 In Vitro Diagnostic Medical Devices Stability Testing)'과 임상 검사실 표준 기구(Clinical and Laboratory Standards Institute, CLSI)에서 발행한 2009년 문서 'EP25A 생체 외 진단 시약의 안정성 평가(EP25A Evaluation of Stability of In Vitro Diagnostic Reagents)'는 모두 생체 외 진단 시약의 안정성을 판단하기 위해 이 방법을 사용할 것을 권장합니다. 다양한 안정성 평가 매개변수는 표 6에 제시되어 있습니다.
표 6. 안정성 평가 매개변수

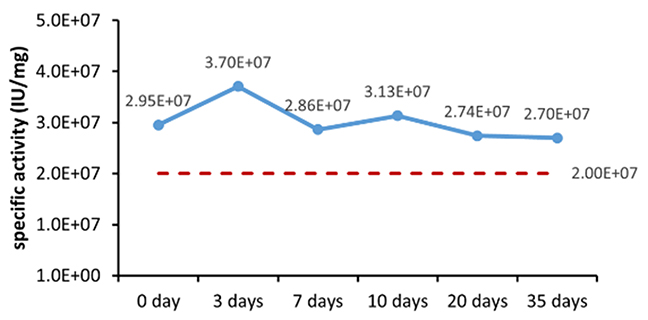
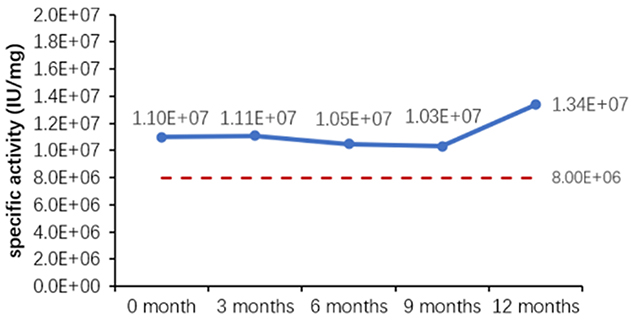
그림 1. 완제품(분말)의 (A)가속 및 (B)실시간 안정성 평가.
GMP 등급 원료 및 시약 제품의 품질 관리 평가 시에는 일반적으로 더 잘 확립되어 있고 민감도가 더 높으며 지정된 요구 사항을 충족하는 평가를 이용합니다. 분석 방법의 잠재적 한계로 인해 원료 물질뿐만 아니라 최종 치료제를 종합적으로 평가하기 위해 상호 보완적인 방법을 동시에 활용하는 것을 고려하는 것이 좋습니다. 분석 방법 자체 또한 엄격한 밸리데이션을 거쳐야 합니다. 잘 확립되어 있으며, 엄격하고 전문적인 품질 관리 시스템은 모든 치료제 승인 절차의 성패를 좌우할 수 있으며, USP와 같은 약전 표준을 참조해야 합니다. 따라서 의약품 품질 관리에 대한 국제적 관점은 '검사를 통한 의약품 품질 관리'에서 '제조 공정 관리를 통한 의약품 품질 달성'으로, 그리고 마지막으로 '좋은 설계를 통한 의약품 품질 확보 (품질 기반 설계(Quality by Design, QbD))'로 발전해 왔습니다. 이러한 QbD 철학을 수용한다는 것은 제품 품질 특성과 임상적 안전성 및 유효성 간의 상관관계를 확립하는 것을 의미하며, 전체 공정 및 제품 수명 주기 관리 전반에 걸쳐 포괄적인 품질 관리 시스템을 구축해야 한다는 것을 의미합니다. 따라서 ACROBiosystems는 이러한 사고방식을 바탕으로 세포 유전자 치료제의 원료 물질을 안정적으로 공급할 수 있도록 GMP 품질 관리 시스템을 지속적으로 개선하고 있습니다.

This web search service is supported by Google Inc.
















 A-Z
A-Z

























