주문제작 서비스 문의
찾으시는 제품이 저희 사이트에 없으신가요? 단백질 주문 제작 서비스를 이용해보세요!
주문제작 서비스 문의 >>





















 Limited Edition Golden Llama is here! Check out how you can get one.
Limited Edition Golden Llama is here! Check out how you can get one.  Limited Edition Golden Llama is here! Check out how you can get one.
Limited Edition Golden Llama is here! Check out how you can get one.
Offering SPR-BLI Services - Proteins provided for free! Offering SPR-BLI Services - Proteins provided for free!
Here come GMP Grade Cytokines!Free Sample is available! Here come GMP Grade Cytokines!Free Sample is available!
> Insights > Pre-formed Fibrils: Another Perspective in Modelling of Neurodegenerative Diseases Neurodegenerative diseases inflict a substantial burden on both personal, societal, and public health across the globe. These progressive neurological disorders feature a set of debilitating, incurable diseases that are increasing in prevalence, such as Alzheimer’s disease (AD) and Parkinson’s disease (PD). Together, both AD and PD afflicts over 130 million people worldwide, with its prevalence rising dramatically with age. This number is only expected to increase in the foreseeable future as life spans continue to increase. Unfortunately, neurodegenerative disease research has been limited by an inadequate understanding or incomplete perspective into disease pathology. This, in turn, has translated into a poor track record in developing neurodegenerative treatments and discovering new therapeutic avenues.
Elucidating disease pathology has been heavily driven through the use of in vitro and in vivo models; both essential tools in providing new insights into disease progression, cellular mechanisms, and much more. In the case of neurodegenerative diseases, our limited comprehension is in part due to the use of premature experimental models.1 Existing neurodegenerative disease models relies on the use of transgenic mouse models with knock-out or over-expressed pathogenic genes and proteins. However, these models often do not completely reproduce the natural disease progression and are an incomplete imitation of neurodegeneration. Pre-formed fibrils (PFFs) are a novel tool for modeling AD and PD which are being increasingly used in related studies. These fibrils are generated in vitro and can imitate endogenous protein aggregation in both animal and cellular models. As such, PFFs enable the creation of a model that more closely aligns with the traits found in neurodegenerative diseases including ‘seeding’ and ‘transmission’ of the related pathogenic proteins. In this article, we first discuss the limitations of current disease models before discussing in-depth about PFFs and its current applications in neurodegenerative research.
A hallmark of neurodegenerative diseases is protein misfolding and aggregation of proteins that results in cellular dysfunction, loss of synaptic connections, and brain damage. Although the protein that aggregates may differ between neurodegenerative diseases, the disease progression and clinical indications are similar. To study these diseases, most of these experimental models are animal models involving the use of transgenic rodent animals expressing human genes to induce pathogenic protein aggregation, especially AD. Similarly, chemically or physically induced animal models are also employed as these models are able to capture certain symptoms of diseases that may not be modelled in transgenic mice, such as PD.
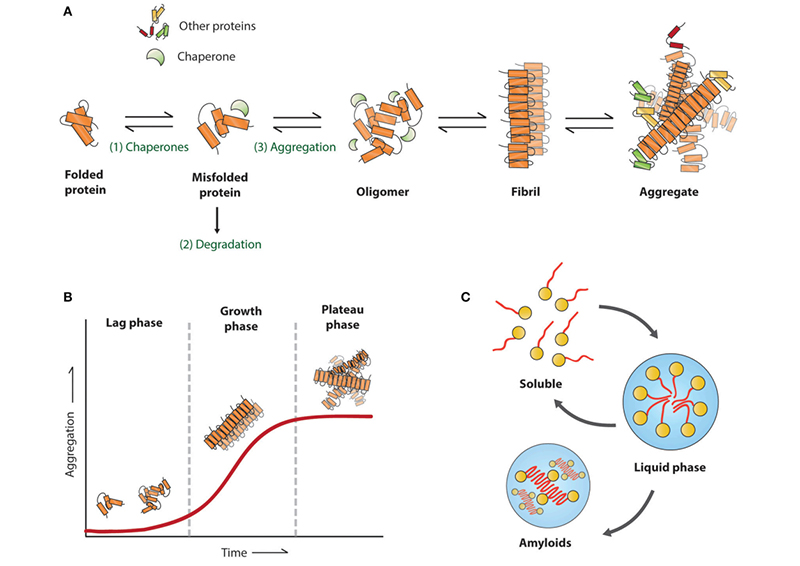
Figure (1). Amyloid aggregation associated with neurodegenerative diseases. Reproduced from Stroo et al. 2
AD disease models use transgenic mice: these models generally only exhibit amyloid accumulation, a defining characteristic of AD, but lack neurofibrillary tangle (NFT) development. NFTs are a secondary characteristic of AD that are insoluble twisted fibers inside of brain cells. These tangles are formed through a misfolded tau protein that are the constituents of microtubules. These microtubules transport nutrients and other molecules between nerve cells; the formation of NFTs causes the collapse of these microtubule structures. Within the transgenic mouse models, endogenous mouse tau inhibits the aggregation of human tau and only express 4R tau isoforms, unlike the mixture of 3R and 4R isoforms in AD. To overcome the non-formation of NFTs, transgenic mice were modified to express 4R human tau with a P301L or P301S mutation. However, this reduces the accuracy of the mouse model, as these mutations are not associated with AD and may potentially influence toxicity or interaction with the amyloid plaques through pathways not reflective of AD. Furthermore, the overexpression of these mutated tau proteins results in motor deficits not associated with AD and interferes with cognitive testing.3
To model PD, toxin-induced mouse models such as 6-OHDA and MPTP, are the classical model choice and are widely in-use. By introducing neurotoxins into the animal models, rapid degeneration of dopaminergic neurons in PD-related regions leads to a robust and well-characterized motor deficit. However, this model only simulates the clinical symptoms and lacks the molecular pathology of PD, such as alpha-synuclein (α-syn) accumulation and Lewy bodies. On the other hand, transgenic mice with mutations in SNCA, LRRK2, Parkin, PINK1 and DJ-1 are also used to study PD. These genetic mutations are linked to inherited forms of PD and express the implicated proteins. However, these transgenic mouse models often lack the same robust nigrostriatal degeneration (loss of dopamine neurons) and do not have the same consistent, reproducible motor deficits as with the toxin-induced models.
Unlike transgenic and toxin-induced mouse models, PFFs provide a different and more cost-effective perspective into neurodegenerative disease research. Whereas transgenic mouse models focus on selecting certain genetic mutations to express pathogenic proteins endogenously, the genetic differences between host and target could cause some complications. This is most notable with the lack of NFT formation due to the inhibition of expressed human tau by endogenous mouse tau. On the other hand, toxin-induced models simulate the clinical symptoms in a reproducible format but lack the molecular pathology needed to understand disease progression. PFFs are a more direct methodology, injecting pre-formed pathogenic fibrils to form protein aggregates and induce neurodegeneration. This method not only induces the molecular pathology faster than transgenic mice, but also reduces the scientific burden and length of time for mouse maturation. Similarly, PFFs are useable at the in vitro level, enabling a direct, more reproducible method to identify and measure disease pathology while providing scientists a model to test new drug candidates.
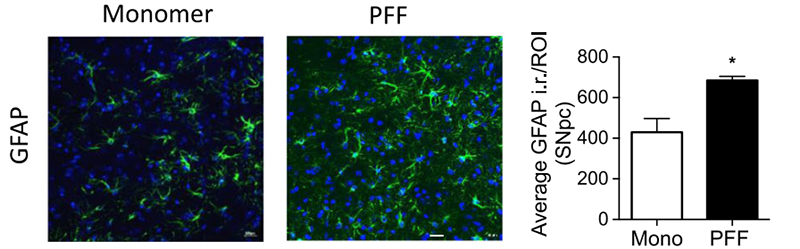
Figure (2). Immunohistochemical GFAP staining of the substantia nigra pars compacta region in a mouse brain. Optical density analysis of GFAP-positive cells reveals an increased amount of astrogliosis in PFF α-syn treated mice in comparison to monomer α-syn, revealing neuroinflammation associated with α-syn accumulation. Reproduced from Earls 2019.4
PFFs are easily formed from protein monomers by incubating at 37°C and shaking with or without heparin. Each protocol depending on the protein monomer of choice depends on the monomers’ ability and tendency to aggregate. For example, tau-441 PFFs require a concentration of 2 mg·mL-1 with heparin, while shaking and incubating for 7 days. In contrast, alpha-synuclein PFFs are incubated without heparin at an initial monomer concentration of 5 mg·mL-1. For other mutated monomers or pathogenic fragments such as alpha-synuclein A53T or tau K18 P301L, incubation time can be much shorter (4-5 days) due to its increased tendency to aggregate.
Verification of PFFs is also critical before implementation as disease models. The presence of fibrillary structures should be visually verified through microscopy and chemically through ThT assays. ThT assays help analyze the structure of high molecular weight species and determine whether the typical cross-beta structure has been formed. Similarly, size and morphology of aggregates should be confirmed through transmission electron microscopy (TEM) or atomic force microscopy. In addition, the length of the formed fibrils is important in pathogenicity. Thus, before use, PFFs should be sonicated into 50 nm or smaller seeds before experiments; both of which can be visualized through TEM or dynamic light scattering.
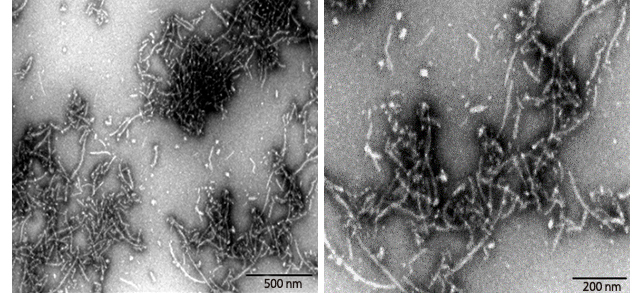
Figure (3). Transmission Electron Microscopy of Human Tau-441/2N4R pre-formed fibrils(ACROBiosystems, Cat. No. #TAU-H5115). Protein aggregates with distinct fibrous structures are visible with accurate morphology.
Tau PFF models for AD study and drug development are well established in cellular systems including neurons, microglia, and astrocytes. Studies evaluating PFF addition to cultured cells have indicated that the tau PFFs are internalized by cells through endocytosis and serve as a seed to recruit endogenous tau to adopt misfolded conformations.5,6 After internalization of tau PFFs, intracellular fibrilization of endogenous tau was induced. After recruitment, tau aggregates are externalized through degenerating axons or somatodendritic compartments via secretion or other undiscovered mechanisms. The externalized tau aggregates are then adopted by surrounding or interconnected neurons, sustaining the theorized template for the propagation of tau pathology. In contrast to cell-based models, animal-based tau PFF models have proven to be more of a challenge to establish. Injection of heparin-induced wild-type tau-441 PFFs were found to have a limited seeding capacity and instead, require the combination of transgenic mice and truncated tau-PFFs to achieve AD-like protein aggregations. In one study, in vivo studies were established through the hippocampal or frontal cortex injection of truncated tau PFFs into tau Tg mice expressing human mutant P301L tau. 7 Through the combination of both transgenic mouse and PFF models, both induction of tau fibrillization and spreading of tau pathology to interconnected brain regions was observed. Neuronal loss in hippocampus was observed, leading to the discovery of a new in vivo model for tau pathological spread with neurodegeneration.
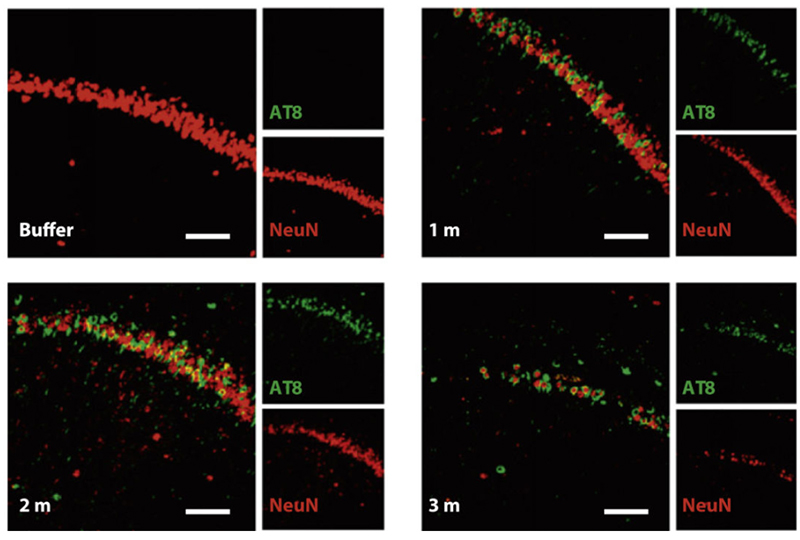
Figure (4). Nissl staining of the hippocampus of P301L mice injected with buffer or truncated tau PFFs. AT8 (green, identifier of tau-presence) was apparent in mice treated with PFFs and shown to preceed neuronal loss found in the 3 month post-injection mice. Reproduced from Peeraer 2015.7
In PD, α-syn PFF models are commonly used to model the two major disease processes: accumulation of intraneuronal Lewy-bodies/neurites and selective degeneration of midbrain dopamine neurons. Whereas tau PFFs struggle to induce protein aggregations in vivo, α-syn PFF models have been well-established in both in vitro and in vivo. In vitro models have been well established targeting primary neurons8, human iPSC-derived dopaminergic neurons9, and organ-on-a-chip10, all of which serve as efficient tools for high-throughput drug screening and testing. Similarly, α-syn PFFs efficiently seed aggregation and fibrillization of endogenous α-syn in wild-type mice. In one study, α-syn PFFs were injected, targeting the dorsal striatum, which resulted in the identification of hyperphosphorylated deposits of α-syn. 11 After 30 days post-injection, the Lewy-body like accumulations travelled along the inter-neural connectivity, suggesting a cell-to-cell transmission method. Clinical symptoms related to PD were also observed, including impaired balance and motor coordination. Overall, a single intra-striatal injection of misfolded α-syn PFFs was sufficient to initiate the neurodegenerative cascade associated with PD, including Lewy-body pathology, dopaminergic neuron loss, and impaired motor function.
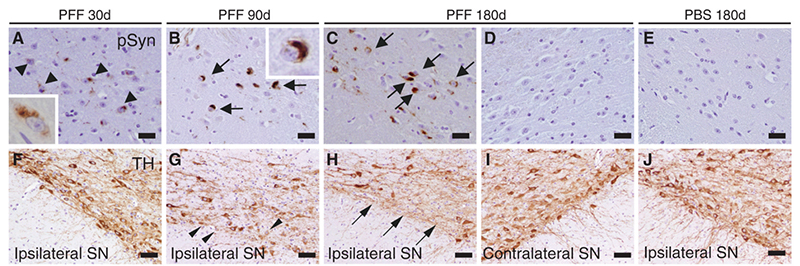
Figure (5). α-Syn immunostaining of substantia nigra pars compacta of mice following intrastriatal PFF injection. Lewy-body like inclusions (black arrows) were observed in ipsilateral sections but were not observed in contralateral sections; this supports the theory of a cell-to-cell transmission method for Lewy-bodies comprised of misfolded α-syn. Reproduced from Luk 2012.11
Overall, pre-formed fibrils are novel tools that model a more ‘natural’ pathophysiology of neurodegenerative diseases, including AD and PD. PFFs mimic the seeding and transmission activity of pathogenic proteins and can be used for both in vivo and in vitro models. As such, compared to transgenic mouse and toxin-induced models, PFFs have been shown to better replicate major neurodegenerative disease hallmarks with less scientific burdens.
Despite PFFs models finding success in academic research, the application of these models is limited in clinical, industry-driven research. Without stable, quantifiable phenotypes as well as reproducible protocols, PFF-based modelling will continue to find a limited application in large-scale drug screening and testing. However, many scientists and industries have dedicated efforts into dissecting the structural features and activity mechanisms of PFFs in disease models. With a deeper understanding of these characteristics, higher quality PFFs can be produced which helps provide more in-depth examinations into neurodegenerative diseases. In turn, PFFs could become the breakthrough modeling platform for scientists to discover and develop the next therapeutic for neurodegenerative diseases.
1. Gitler AD, Dhillon P, Shorter J. Neurodegenerative disease: models, mechanisms, and a new hope. Dis Model Mech. 2017; 10(5):499-502
2. Stroo E, Koopman M, Nollen EA, Mata-Cabana A. Cellular Regulation of Amyloid Formation in Aging and Disease. Front Neurosci. 2017; 14;11:64.
3. Drummond E, Wisniewski T. Alzheimer's disease: experimental models and reality. Acta Neuropathol. 2017; 133(2):155-175.
4. Earls, R.H., Menees, K.B., Chung, J. et al. Intrastriatal injection of preformed alpha-synuclein fibrils alters central and peripheral immune cell profiles in non-transgenic mice. J Neuroinflammation. 2019; 16, 250.
5. Frost B, Jacks RL, Diamond MI. Propagation of tau misfolding from the outside to the inside of a cell. J. Biol. Chem. 2009; 284:12845–12852.
6. Guo JL, Lee VM. Seeding of normal Tau by pathological Tau conformers drives pathogenesis of Alzheimer-like tangles. J. Biol. Chem. 2011; 286:15317–15331.
7. Peeraer E, Bottelbergs A, Van Kolen K, et al. Intracerebral injection of preformed synthetic tau fibrils initiates widespread tauopathy and neuronal loss in the brains of tau transgenic mice. Neurobiol Dis. 2015; 73:83-95.
8. Volpicelli-Daley LA, Luk KC, et al. Exogenous α-synuclein fibrils induce Lewy body pathology leading to synaptic dysfunction and neuron death. Neuron. 2011; 72(1):57-71.
9. Tanudjojo, B., Shaikh, S.S., Fenyi, A. et al. Phenotypic manifestation of α-synuclein strains derived from Parkinson’s disease and multiple system atrophy in human dopaminergic neurons. Nat Commun. 2021; 12, 3817.
10. Pediaditakis I, Kodella KR, Manatakis DV, et al. Modeling alpha-synuclein pathology in a human brain-chip to assess blood-brain barrier disruption. Nat Commun. 2021;12(1):5907.
11. Luk KC, Kehm V, Carroll J, et al. Pathological α-synuclein transmission initiates Parkinson-like neurodegeneration in nontransgenic mice. Science. 2012; 338(6109):949-53.

This web search service is supported by Google Inc.
















 A-Z
A-Z























