























 Limited Edition Golden Llama is here! Check out how you can get one.
Limited Edition Golden Llama is here! Check out how you can get one.  Limited Edition Golden Llama is here! Check out how you can get one.
Limited Edition Golden Llama is here! Check out how you can get one.
Offering SPR-BLI Services - Proteins provided for free! Offering SPR-BLI Services - Proteins provided for free!
Here come GMP Grade Cytokines!Free Sample is available! Here come GMP Grade Cytokines!Free Sample is available!

Given the aseptic nature of cell therapy product production, it is imperative that the raw materials used in the process be sterile. The failure to ensure sterility represent a critical setback in cell therapy product manufacturing, leading to the inability to deliver drugs that meet quality requirements to patients in a timely manner.
Hence, sterility stand out as is a pivotal quality attribute for raw materials in the context of cell therapy. The aseptic control strategy employed during the raw material production process assumes heightened signficance, especially for the production processes of cytokines, antibodies, and enzymes. These processes often entail non-terminal sterilization processes. Conducting a thorough risk assessment is critical to identify potential sources of microbial contamination . The assessment should encompass a comprehensive examination of various factors, including, but not limited, to the facility, equipment, process design, material handling, personnel requirements, production operations, and environmental monitoring. Subsequently, it is essential to formulate targeted control measures to enhance both production management and quality control.
In this context, aseptic control strategies are subject to strigent standards set forth by various Good Manufacturing Practice (GMP) regulations and guidelines across different countries. The industry consistently adheres to a set of commonly recognized GMP regulations and guidelines, encompassing:
1. European Union Good Manufacturing Practice (EU GMP) Annex 1: Manufacture of Sterile Medicinal Products
2. United States FDA current Good Manufacturing Practice (21 CFR Part 210, 211, 600)
3. FDA Guidance for Industry: Sterile Drug Products Produced by Aseptic Processing
4. PIC/S GMP Annex 1: Manufacture of Sterile Medicinal Products
Among them, the new version of Annex 1 of EU GMP, which was introduced on August 25, 2023, has the most stringent requirements for the production of sterile products. The regulation specifically covers active pharmaceutical ingredients, excipients, primary packaging materials, finished dosage forms, as well as various packaging sizes, production processes, and technologies. Meanwhile, the regulation provides general guidance on the overall design and control of facilities, equipment, systems, and procedures used for the production of all sterile products, following the principles of quality risk management (QRM). The aim is to ensure the absence of microorganisms, particles, and endotoxin/pyrogen contamination in the final product. The guidance emphasizes the overall assessment and contamination control from aspects such as facility, equipment, process, material, testing, and environmental monitoring.
ACROBiosystems ensures the compliance of its GMP-grade cell therapy products with the aforementioned regulatory and guidance standards. Employing an integrated approach, the company meticulously designs and incorporates aseptic control strategies for facilities, equipment, materials, processes, and personnel. Continuous refinement and enhancement are integral to ACROBiosystems' commitment to quality assurance, facilitated by the systematic application of PDCA (Plan-Do-Check-Act) tools

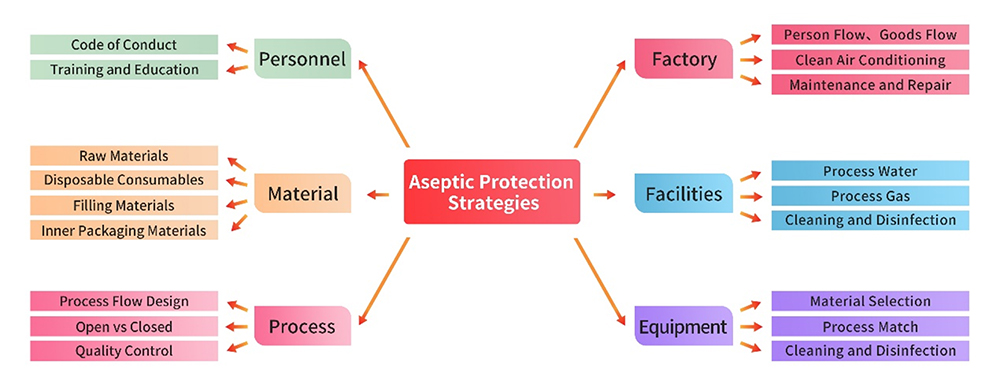
1. Facilities and Equipment
The production facility, utilities, and equipment adhere to strigent GMP regulations. Prior to initiating product production, a comprehensive gap analysis and risk assessments are conducted for each facility and piece of equipment. Written procedures are developed and strictly enforced, covering areas such as usage, cleaning, disinfection, and maintenance. These measures ensure that the management of these infrastructure is executed with precision, preventing any adverse impact on aseptic control.
2. Personnel Management
Personnel engaged in production and quality management activities possess the requisiteeducational background, training, and experience to uphod both the standard of production and effective control of contamination and cross-contamination.This commitment to personnel qualification ensures a seamless and reliable execution of tasks critical to maintaining the integrity of the production process.
3. Material Management
Raw and auxiliary materials used in production are preferably sourced as pharmaceutical-grade materials, including packaging materials. Stringent supplier management practices are implemented through processes such as qualification collection, on-site audits, etc.Quality control strategies are thoughfully formulated based on comprehensive risk assessment, particulary in the realm of microbiological control, ensuring the integrity of the materials used in the production process.
4. Production Process and Quality Control
Key points for aseptic control in the production process include:
| Production Environment |
Raw Material Preparation: ISO Class C and A Drug Product Production: ISO Class B and A Dynamic environment monitoring |
| Solutions and Process Gases |
0.1μm / 0.22μm sterilizing filtration Drug product production with secondary sterilizing filtration Integrity testing before and after filter use |
| Single-Use Consumables |
Gamma irradiation sterilization Integrity check before use |
| Process Control |
Aseptic connections Bacterial endotoxin control Aseptic process simulation filling |
| Equipment Control | Cleaning processes and cleaning confirmation for reusable equipment |
| Cleaning and Disinfection | Disinfectant efficacy verification |
| Quality Control |
Microbial limits control for raw materials Sterility testing of products complies with USP |
Upstream Cell Cultivation:
• In the cell culture production phases, strict regulation govern personnel gowning and behavior requirements to minimize contamination risks from operations.
• Cell recovery and flask expansion stages are conducted in a Class C clean area as the background environment.Open operations are performed in a Class A biosafety cabinet under laminar protection withdynamic environmental monitoring occuring simultaneously. To ensure aseptic conditions, reactor expansion and perfusion culture stages incorporate disposable closed systems, employing sterile welding technology for process like inoculation, feeding, and other processes to effectively avoid microbial contamination.
• During the cell culture stage, materials used undergo rigorous endotoxin control and are only used after passing stringent quality release criteria. The preparation of culture media and solutions adheres to the pharmacopoeial requirements for injection water. After preparation, the solutions are sterilized by passing through 0.1μm/0.22μm filters, and the integrity of the filters is tested. Simultaneously, sterility testing is performed, and solutions can be proceed to cell culture production only if both tests yield satisfactory results. Gases used in the process, such as carbon dioxide, oxygen, and compressed air, maintainhigh-purity or food-grade standards. Process gases connected to the reactor undergo effective sterilization by passing through a 0.22μm filter. Disposable consumables, including reaction bags and storage bags, undergosterilization by radiation to ensure sterility, ensuring that materials in direct contact with the liquid remain uncontaminated by microorganisms.
• Deep filtration membranes used for clarification filtration are for single use. Before use, membrane packages are rinsed with injection water to remove impurities contained in the membrane packages. Following clarification filtration, the liquid undergo a second filtration through a 0.22μm filter to effectively reduce microbial load before entering downstream purification processes. Prior to transitioning to downstream purification, the harvested clarified liquid undergoes endotoxin testing to ensure the intermediate product meets the process requirements.
Downstream purification:
• The separation columns and chromatographic media employed in the purification process are exclusively dedicated to the specific project, preventing cross-contamination risks across different production stages. Post each batch production, the chromatographic system undergoes a thoroguhcleaning, confirmed for cleanlines based on risk assessment. Chromatographic media, following written procedures, are cleaned and stored appropriately, undergoing a subsequent cleaning before use in the next batch production, effectively mitigating the impact of microorganisms.Microbial control methods for materials and consumables in direct contact with products during the purification stage mirror those in the cell culture stage, ensuring that introduction of microorganisms is prevented. Reusable equipment and instruments are sterilized by moist heat to maintain sterility.
• The liquid post-separation and chromatography undergoes nanofiltration and ultrafiltration processes, culminating in sterilizing filtration using a 0.2μm filter. Following filtration, the liquid is filled under grade A laminar flow protection to reduce microbial contamination from the environment. The bulk liquid undergoes various tests, including microbial limits and endotoxin assessments, in adherence to the quality standards. Upon passing these tests and receiving approval from quality department, the liquid proceeds to final product filling.
Formulated product:
• The filling of the finished product follows a non-terminal sterilization aseptic production process, ensuring a strict B+A production environment. Post-sterilizing filtration, the semi-finished product is acquired. Assembly, filling, capping, automated loading and unloading, and lyophilization are performed using fully automated equipment and single-use aseptic filling systems within an grade A aseptic environment (PMS continuous environmental monitoring system). Following lyophilization, sealing is completed in a C+A environment. The packaged products undergo manual visual inspection, labeling, and additionaloperations. They are only placed into finished product storage and released post-inspection.
• Rigorous aseptic process validation includes regular confirmation of aseptic process control, incorporating the use of aseptic nutrient media and/or product substitutes in APS (media simulation vials). APS evaluates all aseptic operations performed from sterilization and cleaning of materials to container sealing. The media undergoes growth promotion testing to ensure reliable culture results. The microbial ingress experiment serves as a means to guarantee the sealing integrity of the product packaging system, ensuring product sterility during storage.
• Finished product quality control: The sterilization test is the final step to ensure sterility among a series of critical control measures. Sterility testing is conducted inside an aseptic isolator, and sampling for sterility testing includes products at the beginning and end of batch filling for representation. Sterility analysis methods strictly adhere to pharmacopoeial requirements, using ATCC and CMCC-authorized strains, and following the pharmacopoeial requirements for culturing biological products to ensure the reliability of results.
In summary, ACROBiosystems' GMP-grade products are produced with comprehensive aseptic protection strategies, stringent quality control, and compliance with GMP regulations. These asepetic processes support the registration and commercialization of CGT products.
• Controlling external contaminants in the production of critical materials for cell and gene therapies.
• Aseptic Protection Strategies for Critical Material Production in CGT
• Quality Control System for Critical Materials in CGT
• Global Supply Chain Security System for Critical Materials in CGT
• How to Better Meet Regulatory Requirements of the United States for Critical Materials in CGT
• ...





This web search service is supported by Google Inc.
















 A-Z
A-Z



















